

Le besoin d’appliquer des statistiques appropriées en génomique a été bien documenté. Plusieurs approches statistiques astucieuses ont été mises au point et de nombreux logiciels ont été développés (certains gratuits et très performants). Malgré tout, je reçois fréquemment la visite d’un étudiant ou chercheur qui se demande s’il doit rapporter la magnitude d’un effet (ex. le gène X est sur-exprimé 4.5 fois dans la condition A vs B) ou le niveau de signification statistique de cette magnitude (ex. le gène X est sur-exprimé dans la condition A vs B avec une p-value de 0.0012). Il faut savoir qu’une p-value est systématiquement liée à un test statistique (test d’hypothèse), ce type de test n’étant qu’une formulation mathématique d’une question précise posée sur les données. Pour obtenir une p-value, cette question prend la forme d’une hypothèse (ex. le gène X n’est pas différentiellement exprimé), appelée $H_0$ (hypothèse nulle) dans le jargon statistique. Une p-value proche de zéro suggère que l’hypothèse n’est pas confirmée (ex. les données ne supportent pas la non-sur-expression du gène X… Remarquez la double négation!). Dans la pratique, cette nuance pose problème dans deux cas: lorsqu’on observe de faibles et inconséquentes différences dans de grands jeux de données OU lorsqu’on observe de grandes différences accompagnées d’un bruit inattendu (ex. outlier). Pour illustrer, voici deux exemples obtenus par simulation (on peut imaginer qu’il s’agit de quantification par qPCR d’un gène dans deux conditions).
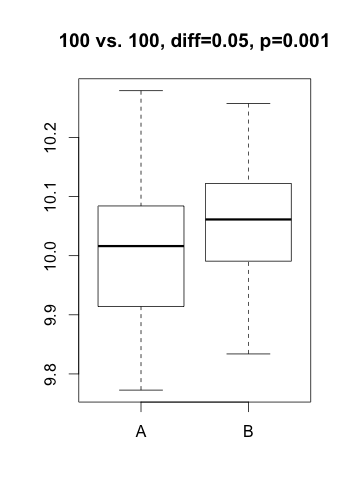

Dans le premier scénario, le test d’hypothèse indique que la différence est statistiquement significative ($p < 0.01$), mais on remarque que la différence d’expression est sans doute inintéressante (augmentation de 3%) et possiblement le résultat d’un biais difficile à déterminer. Dans le second scénario, l’un des trois échantillons de la condition « B » se comporte étrangement et résulte en un test non-significatif malgré une différence substantielle. Dans les deux cas, la différence dans les moyennes d’expression aurait donné une meilleure indication de la situation. Dans la majorité des projets dans lesquels j’ai été impliqué (puce à ADN, RNA-Seq, criblage haut-débit, protéomique, qPCR, etc.), ma première suggestion est toujours de débuter l’analyse en utilisant la magnitude de l’effet sous étude (différence, log-ratio, %inhibition, etc.). De manière générale, je recommande de se demander si l’hypothèse du test envisagé correspond bien à une question scientifiquement pertinente (ex. est-ce que les conditions A et B influencent l’expression du gène X?). Je vous promets de revenir dans un prochain article avec de bons exemples d’application de tests d’hypothèse… D’ici là, n’hésitez pas à laisser vos commentaires!
Laisser un commentaire