

Suite à l’analyse de votre échantillon par spectrométrie de masse, vous recevez généralement vos résultats sous forme d’une liste de protéines. Lors du traitement des données, certains facteurs influencent quelles protéines se retrouveront dans la liste finale.
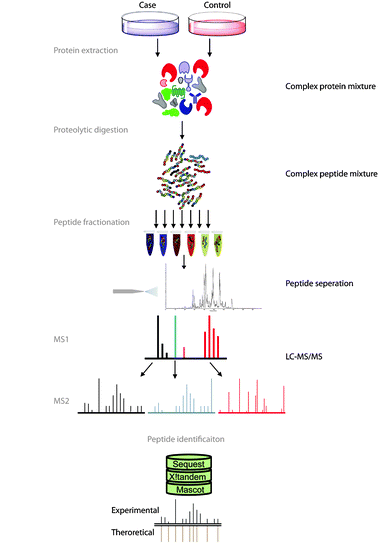
Fig. 1 Approche ascendante. Figure modifiée provenant de Angel et al. (2011)
Commençons d’abord par expliquer brièvement comment cette liste de protéines est obtenue. L’approche de protéomique ascendante est celle qui est couramment utilisée (voir Figure 1). Dans cette approche, l’extrait protéique est d’abord digéré avec une protéase (généralement la trypsine) afin de créer un mélange de peptides qui sera injecté sur le système de chromatographie liquide couplé au spectromètre de masse.
L’instrument fera ensuite l’acquisition de quelques milliers de spectres de masse de fragmentation. Ces spectres seront d’abord utilisés pour identifier des peptides. Ces peptides seront à leur tour utilisés pour déduire la liste des protéines.
Une approche efficace pour associer une séquence peptidique à un spectre de masse est d’utiliser un logiciel qui fait la recherche des masses observées dans une base de données de séquences de protéines. Le choix de cette base de données est donc crucial pour identifier les protéines contenues dans l’échantillon. Si une protéine est absente de la base de données, elle ne peut évidemment pas être identifiée! Voici quelques conseils pour bien choisir la base de données.
- Le premier critère pour sélectionner la base de données est de choisir d’abord l’organisme correspondant à l’échantillon.
- Ensuite, il faut considérer s’il est nécessaire que cette dernière contienne les séquences des isoformes, protéines hypothétiques, mutations ou encore la séquence d’une protéine synthétique.
- Un dernier point à considérer est que les identifiants des séquences permettent la récupération d’annotation de diverses bases de données. Ceci est un point important pour les analyses bio-informatiques subséquentes.
Une fois la recherche complétée avec la base de données sélectionnée, l’outil de recherche retourne une liste de peptides. Pour chaque paire spectre-peptide, un pointage indique si le patron de fragmentation contenu dans le spectre correspond bien à la séquence du peptide. L’utilisation combinée d’une base de données leurres lors de la recherche permet d’estimer le taux de faux positifs pour un certain seuil du pointage peptidique. La taille finale de notre liste de protéines identifiées est donc dépendante de notre tolérance pour les faux positifs.
Une fois que le logiciel a généré une liste de peptides avec l’ensemble de spectres, il faut finalement inférer la liste des protéines. Pour illustrer ce problème, imaginez que l’on tente de compléter simultanément plusieurs casse-têtes dont il nous manque des pièces et dont certaines sont identiques entre les casse-têtes. Selon les évidences peptidiques, il est soit possible de confirmer la présence d’une protéine (peptides uniques) ou la présence d’un groupe de protéines (peptides communs).
Le principe du rasoir d’Ockham ou principe de parcimonie est employé pour construire une liste minimale de protéines. Selon ce principe, l’algorithme assigne itérativement le plus gros sous-ensemble de peptides restant à une protéine. Un problème relatif à cette approche est qu’il arrive fréquemment qu’il sélectionne des protéines hypothétiques ou pauvres en annotations (le choix de votre base de données peut mitiger ce problème). Bien souvent aussi, notre protéine d’intérêt ne se retrouve pas dans la liste générée par le principe d’Ockham malgré que plusieurs peptides communs soient identifiés. Dans ce cas, il est nécessaire de vérifier si elle est présente dans la liste complète des protéines possibles.
Pour ajuster le niveau de confiance des protéines contenues dans la liste finale, deux filtres sont communément appliqués. Une protéine est retenue dans la liste finale si au moins x peptides sont identifiés (x = 3 est souvent utilisé en pratique). On peut aussi filtrer selon le pointage d’une protéine qui est une combinaison du nombre de peptides et de leur score respectif.
Finalement lorsque vous interprétez la liste de protéines obtenue, vérifier les points suivants selon le cas pour évaluer le niveau de confiance de votre protéine.
- Quel est le taux de faux positifs sur l’ensemble des peptides identifiés?
- Vérifier pour une protéine d’intérêt si l’ensemble des peptides identifiés contient des peptides uniques à cette protéine avec un pointage peptidique suffisamment élevé.
- Dans le cas d’un isoforme identifié, il y a-t-il un peptide unique couvrant un exon spécifique qui confirme cet isoforme?
- Vérifier si la protéine listée n’est pas seulement la représentante d’un groupe de séquences de protéines.
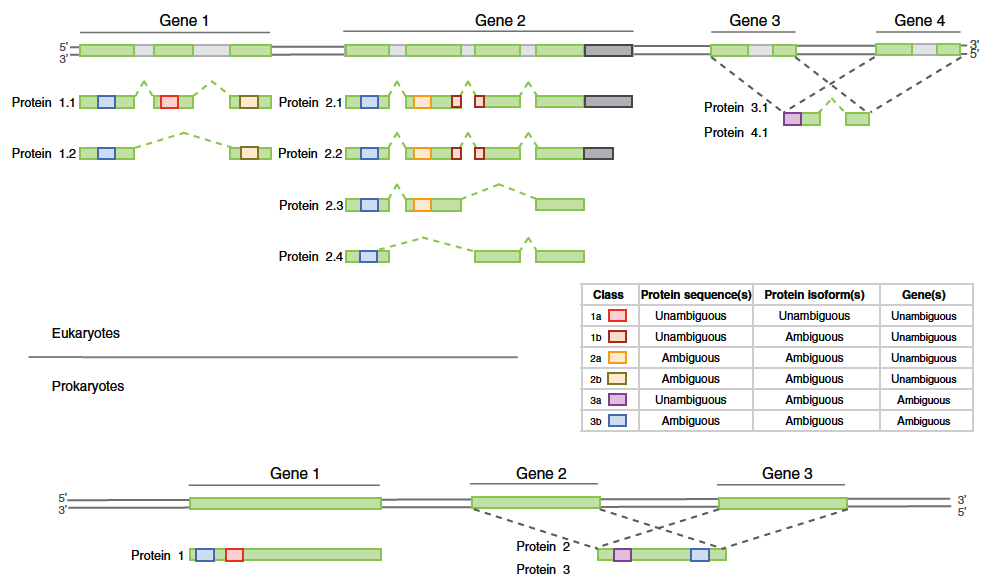
Fig. 2: Survol de l’approche pour mettre en évidence des classes distinctes de peptides chez les eukaryotes et prokaryotes. Figure provenant de Queli et al.(2010)
Bref, il est important de retenir que l’approche protéomique ascendante, ne permet pas une identification absolue de toutes les protéines puisque nous ne disposons pas toutes les pièces du casse-tête et que certaines sont partagées.
Laisser un commentaire