

Cet article est la suite de mon précédent article sur les mégadonnées. Il n’a pas engendré de discussion virtuelle enflammée sur le sujet, mais j’étais très heureuse de recevoir quelques commentaires sur la situation dans d’autres domaines de la bio-informatique.
Protéomique
Mathieu Courcelles, bio-informaticien à la plate-forme de protéomique, explique que la protéomique utilisant la spectrométrie de masse a toujours généré des « mégadonnées ». Donc, l’expression n’est pas très utilisée dans le domaine puisqu’elle n’a rien de nouveau.
Comme il le dit (je me suis permise de traduire ses propos – voir la version anglaise pour la citation originale) :
Les spectromètres de masse sont des instruments qui génèrent un large volume de données 24/7. Très tôt et pour suivre l’évolution des instruments, de l’informatique distribuée a été nécessaire pour processer toutes ces données. J’ai l’impression que le traitement de larges quantités de données n’est pas une nouvelle tendance dans notre champs de recherche. Le mouvement « mégadonnées » nous offre, cependant, un accès pratique à plus de ressources informatiques (nuage informatique) et à un ensemble d’outils génériques pour faire du traitement de données (par exemple, Hadoop, Spark, Dask, Docker). Pour l’instant, il n’y a que quelques laboratoires qui utilisent ces nouveaux outils de traitement de données.
Des processus optimisés et flexibles sont déjà en place pour traiter les données brutes pour identifier les protéines. Toutefois, ces nouveaux processus génériques seront peut-être utiles pour approfondir le traitement des données ou permettre leur intégration. Le partage de données devient aussi populaire en protéomique. ProteomeXchange et HUPO, principaux projets supportés par la communauté, tentent de définir des standards de partage. L’intégration de données est aussi un défi. Un des problèmes particulier est lié aux données manquantes. Tout dépendant des conditions expérimentales et des instruments, la couverture du protéome peut différer grandement.
Données CRISPR
Concernant les données CRISPR, Jasmin Coulombe-Huntington, post-doc au laboratoire de Mike Tyers, est aussi d’avis que le traitement de données massives dans notre domaine existait bien avant l’apparition du mot « mégadonnées ». En traitant les données de screens CRISPR, il fait face aux mêmes problèmes qu’en génomique, principalement ceux liés à l’intégration efficace de données provenant de différentes expériences. Même si, dans son cas, le problème n’est pas lié à l’intégration de données provenant de différentes technologies, la quantité de données générées pose un problème au niveau de la logistique et des ressources. Il croit toutefois que l’intégration est importante, ayant démontré dans ses travaux que l’intégration de différents expériences permettait de mieux contrôler les biais et d’augmenter la valeur de chacune des expériences en permettant de repérer celles qui sont informatives facilement.
Génomique
Dans mon dernier article, j’ai parlé brièvement de Genomic Data Commons (GDC), dont la mission est d’harmoniser les données servies par le site. Chaque échantillon est reprocessé de la même façon pour rendre tous les échantillons comparables. À la plate-forme, nous avons aussi reprocessé les échantillons de TCGA-AML et ceux du projet Leucegene en utilisant un pipeline identique (kallisto pour calculer des TPMs) pour les rendre comparables. La corrélation entre la moyenne d’expression (en log) des gènes entre les deux cohortes est plus grande pour les gènes plus fortement exprimés (les valeurs d’expression pour les gènes faiblement exprimés étant plus bruitées), mais reste globablement bonne. La clé ici est l’utilisation de kallisto qui est beaucoup plus rapide et moins exigeant que les outils d’alignement traditionnels.
D’autres s’engagent aussi dans la voie de l’harmonisation des données. Xena Browser, de UCSC, héberge les données publiques de plusieurs différents projets (GTEx, ICGC, TCGA, TARGET, même les données de projets de perturbation de génome comme Connectivity Map ou NCI ou CCLE pour n’en nommer que quelques-uns) et offre des outils interactifs pour explorer les données. Pour l’une de leurs sources de données, ils ont réanalysé les échantillons de GTEx, TCGA et TARGET en utilisant le même pipeline (kallisto et star/rsem)
pour ainsi permettre une comparaison directe et retirer tous les biais computationnels.
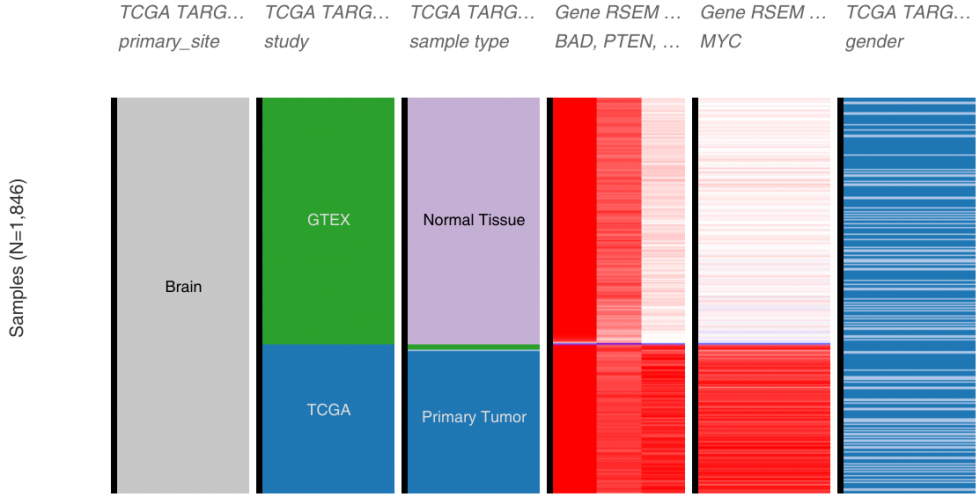
Leur article publié dans Nature Biotechnology, Toil enables reproducible, open source, big biomedical data analyses, décrit leur approche et leurs infrastructures. Cela ne corrige pas les biais expérimentaux ni ceux liés aux échantillons comme tel. Et cela ne pourrait pas servir à intégrer des données provenant de différentes technologies. Toutefois, cela montre que la communauté essaie de trouver des solutions aux problèmes d’intégration.
Voici une bonne suite à notre discussion:
http://computationalproteomic.blogspot.ca/2018/03/big-data-is-not-only-fancycatchy-name.html#more