

This post follows my previous post on big data. Even though the latter did not result in a big virtual discussion, I was pleased to read some comments regarding the situation in other areas of bioinformatics.
Proteomics
Mathieu Courcelles, bioinformatician at the proteomics platform, explained that mass-spectrometry driven proteomics has always generated ‘big data’, so this expression is not used in the field. As he said,
Mass spectrometers are indeed instruments that generate a large volume of data 24/7. Early on and as mass spectrometer instruments evolved, distributed computing was a requirement to process all this data. I feel that processing large amount of data is not a new trend in our field of research. The “Big data” movement is offering us, however, convenient access to more computing resources (cloud) and to new generic data processing workflows (e.g. Hadoop, Spark, Dask, Docker). Right now, there are just a few laboratories that are using these new data processing workflows. Optimized and scalable workflows were already in place for processing raw data to identify proteins. These new generic processing workflows might be handy for further dataset processing or integration. Data sharing is rising up in proteomics. ProteomeXchange and HUPO proteomics standards initiative are the two main projects supported by the community. Data integration is also challenging in proteomics. One particular issue is with missing data. Depending on the experimental conditions and instruments, proteome coverage can differ widely.
CRISPR data
Regarding CRISPR data, Jasmin Coulombe-Huntington, post-doc in Mike Tyers’s laboratory, also thinks that bioinformaticians were processing huge amounts of data before the apparition of the expression “big data”. Dealing with CRISPR screens data, he is facing more or less the same issues as for genomics, mainly regarding efficient integration between different experiments. While the problem is not necessarily integrating data from different technologies, the amount of data makes it a logistic and resources problem. He thinks integration is nevertheless important and has shown in previous work that it can help with better control biases and enhance the value of each screen by making it easier to discriminate the informative from the non informative screen.
Genomics
In my last post, I spoke briefly about GDC whose mission is to harmonize the data they serve by reprocessing raw data in the same fashion so samples can be compared.
At the platform, we have reprocessed samples from TCGA-AML and the Leucegene project using an identical pipeline (kallisto to compute TPMs) in order to be able to compare them. The correlation between the average gene expression (log transformed) in both cohorts was higher for highly expressed genes (lower expressed genes being more noisy) but it was still good overall. The key here was using kallisto, which is much faster and lighter than standard alignment-based tools.
Others are adopting the harmonizing trend. UCSC’s Xena Browser serves publicly available datasets from a number of different projects (GTEx, ICGC, TCGA, TARGET, even data from genome perturbation studies such as Connectivity Map or NCI or CCLE – to name just a few) and provide interactive tools for data exploration. For one of their data hub (data source), they have re-analysed the samples from GTEx, TCGA and TARGET using the same pipeline (kallisto and star/rsem) to allow direct comparison and remove computational biases.
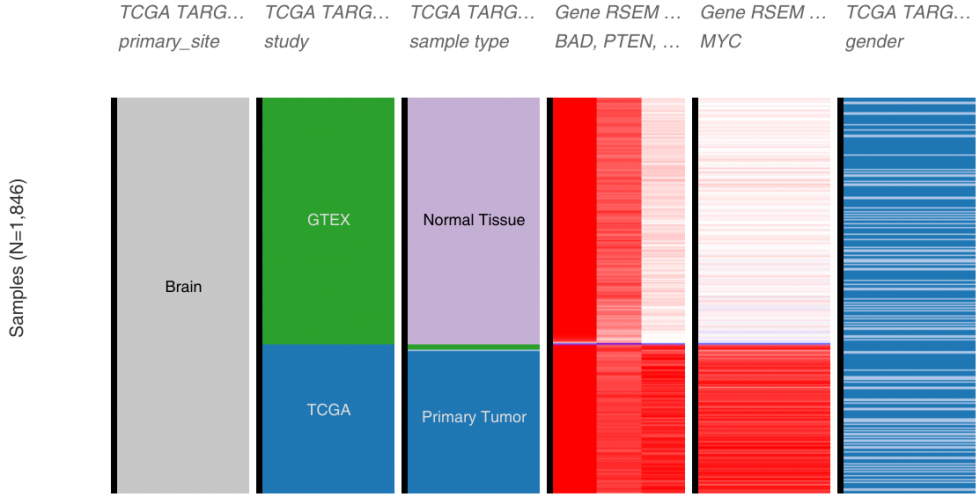
Their paper published in Nature Biotechnology, Toil enables reproducible, open source, big biomedical data analyses, describes their pipeline and infrastructure. This does not correct for experimental and sample handling biases nor could be applied to datasets generated with other technologies, but it shows that the community is trying to deal with the integration problem.
Leave A Comment